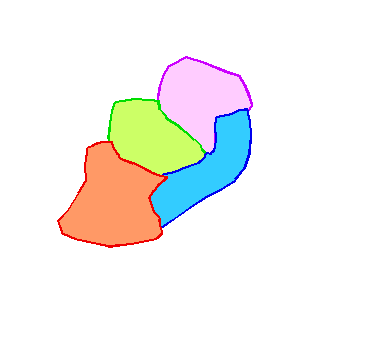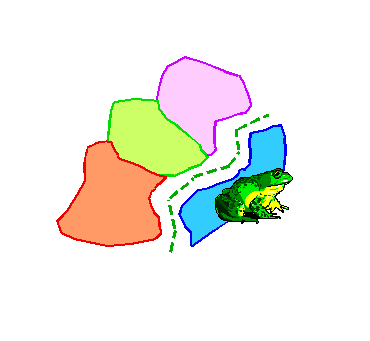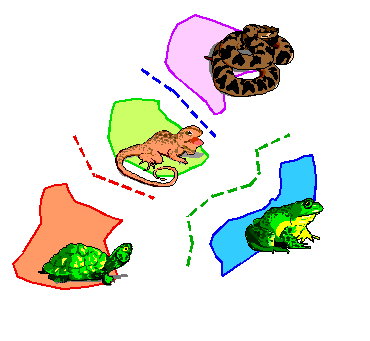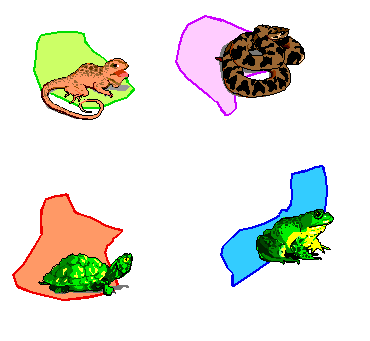
These area cladograms are hypotheses of historical relationships between areas and are derived from phylogenetic and distributional information of the monophyletic groups concerned.
A first-order explanantion for correspondence between phylogenetic relationships of taxa and historical relationships among areas is vicariance.
When formation of barriers or splitting up of areas triggered speciation (i.e. vicariance; as indicated above), all species are endemic to their own area. In such simple cases, derivation of an area cladogram is trivial. Replacement of taxa in the taxon-cladogram by their areas of distribution results in area cladograms with an own and unique terminal node for each area.
However, real data are mostly the result of other processes such as extinction of species in part of their range or dispersal of species over the formed barriers.
As a result of these processes, taxa of a monophyletic groups can become widespread or sympatric in their distribution. In order to obtain area cladograms with an own and unique terminal node for each area, additional steps are necessary.
In vicariance biogeography these additional steps are implemented in three assumptions about the cause of widespread and sympatric taxa. In this PhD project, I compared the implementation of the three assumptions under different a priori and a posteriori methodologies.
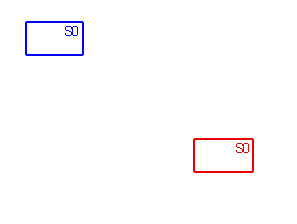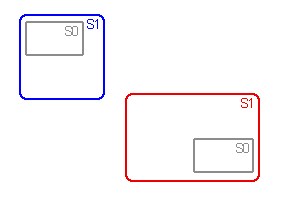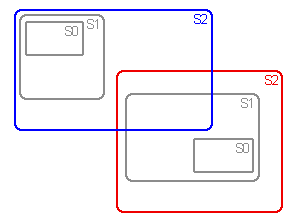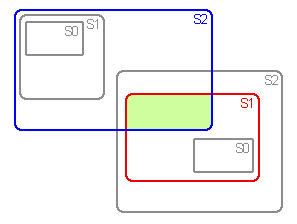
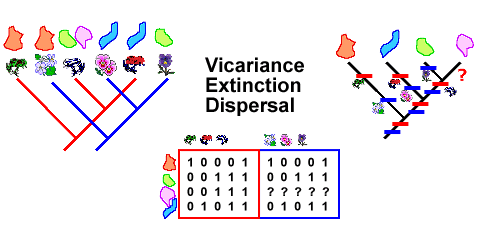
Assumptions zero (A0), 1 (A1) and 2 (A2) are used in vicariance biogeography
to deal with widespread and sympatric taxa.
Sets of area cladograms (solution sets: S0, S1 and S2) are obtained under
these assumptions by using information on phylogenetic relationships between
taxa in its most (under A0) or least (A2) strict sense.
Thereby, different processes are assumed as explanantion for the distributions
of taxa.
Under A0 only the process of vicariance is assumed a priori as explanation
for the widespread or sympatric taxa in a monophyletic group. Under A1
besides vicariance also extinction of taxa in part of their range is a
priori assumed. Under A2, vicariance, extinction and dispersal are assumed
a priori as explanantions for widespread or sympatric taxa.
The processes a priori assumed under A0, A1 and A2 are independent in
their effect on the inference of area cladograms. Therefore, at least
the same area cladograms as already have been derived under a strict assumption
(e.g. A0) should be derived under a less strict assumption (e.g. A1) too.
In other words: the solution sets obtained under A0, A1 and A2 should
be inclusive.
By the use of assumptions that result in inclusive solution sets, general
area cladograms (general solutions) for two or more monophyletic groups
of taxa can be revealed by comparison of solution sets derived under the
same assumption for the separate monophyletic groups.
Results of this PhD project are presented in the following poster:

When the number of taxa and areas is not too big, solution sets can be derived under A0, A1 and A2 by hand. However, since the number of possible area cladograms grows exponentially when more areas are present in a dataset, computer facilities are necessary for derivation of area cladograms in most of the biogeographical studies.
Various authors have implemented A0, A1 and A2 in different programs that enable the researcher to derive area cladograms via different methodologies: CAFCA (Component Compatibility Analysis), PAUP (Brooks Parsimony Analysis), Component 2.0 (Reconciled Tree Analysis), TAS (Three Area Statement Analysis) and Component 1.5 (Component Analysis).
To the left, some of the programs are indicated with links to the pages with information on these programs or their authors.

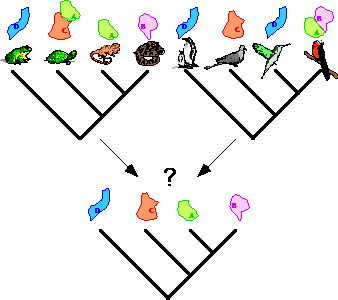
In vicariance biogeography, area cladograms are derived from both distributional
and phylogenetic data of taxa of a particular monophyletic group. These
"single group based" area cladograms give a hypothesis of the history
of distribution of the taxa in this group over a particular set of areas.
It is assumed that speciation within this monophyletic group and break-up
of areas are historically associated and therefore cladogenetic relationships
between taxa can be used to reveal historical relationships between areas.
An area cladogram derived for a single monophyletic group thus gives a
hypothesis on historical relationships between areas in which taxa of
this monophyletic group can be found. However, since only one group of
taxa is used, this hypothesis is only valid with respect to this single
group. In order to obtain a more general hypothesis, an area cladogram
has to be based on data of more monophyletic groups. Such an area cladogram
is called a general area cladogram.
General area cladograms are thus based upon phylogenetic and distributional
data of two or more monophyletic groups of taxa, present in the same set
of areas. To derive these area cladograms, two different categories of
methods can be used: a priori and a posteriori methods. These two categories
of methods differ in the way the obtain general area cladograms.
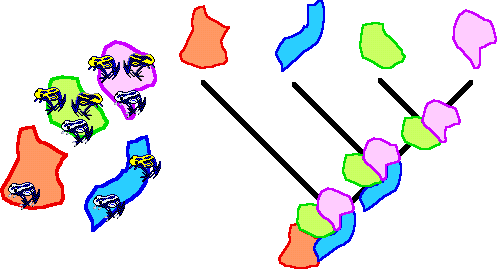
A priori methods derive solution sets under A0, A1 and A2. By comparison
of solution sets derived under the same assumption for different monophyletic
groups, general area cladograms are found in the intersection.
A posteriori methods derive matrices under A0 in which the data of all
monophyletic groups are combined. By a parsimony analysis on this matrix,
general area cladograms are derived.
Results of this PhD project are presented in the following poster:
The geographical distributions of species from different groups are combined into a binary area by taxon matrix. To this matrix a hypothetical outgroup is added for polarization of the data. After application of a standard maximum parsimony analysis, area cladograms of minimal length (number of steps) are derived. Areas in these area cladograms that are grouped together are interpreted as areas between which biotic interchange has occurred.
In this project, the a priori assumptions of PAE were examined and the methodology of this method for inference of area relationships was criticized. This project was performed in close collaboration with prof. dr. D.R. Brooks of the Zoological Department (University of Toronto).
A cooperative project with Cynthia Nolten and Ruud van den Bos.
Adult humans have the ability to 'read' the intentions behind the other's
behaviour as well as to have insight into their own intentions. In other
words, they have meta-representations. This ability is referred to as
Theory of Mind or ToM.
This ability appears to be dependent on the integrity of a neural network
comprised to the (orbito)frontal cortex, amygdala and parts of the temporal
lobe (F-A-T network). ToM and the F-A-T network develop in childhood.
Damage to this network during development leads to the developmental disorder
autism, which is characterized by the inability to read the intensions
behind the other's behaviour.
Greater apes such as chimpanzees do show evidence of ToM, whereas monkeys
such as rhesus monkeys and vervet monkeys lack ToM. Furthermore, within
the great ape clade a development of ToM seems to have taken place. The
monkey-great ape ToM watershed and the great ape ToM development suggest
that the F-A-T network may have evolved in the great ape clade and undergone
subtle changes within this clade.
This project aims at a phylogenetic reconstruction of this F-A-T network
and at comparing this phylogeny with the known phylogeny of non-human
primates and with data on the development of ToM.
Results of this project are presented in the following poster:
- Phylogenetic reconstruction of a neural network underlying Theory of Mind in primates
- Derivation data matrix with characters and character states
- Cladograms and Discussion and Conclusions
This MSc project was performed by mrs. C. Nolten who was supervised by dr. R. van den Bos of the Animal Welfare Centre (Utrecht University) and me.
Phylogenetic biogeography is based on the assumption that cladograms produced by phylogenetic systematic analysis are hypotheses of speciation events. Following this assumption, species cladograms are used not only in studies of species but also in studies of multi-species associations (like several monophyletic groups that are associated in a biogeographical context). Thereby, the evolutionary perspective is that evolution has been historically very contingent and complex. As a consequence, for robust inference of evolutionary processes in a geographical context, analyses of both common and unique patterns are necessary, thereby explaining both congruent and incongruent data.
Phylogenetic biogeography is associated with a posteriori methods. A posteriori methods do not allow any a priori modification on species cladograms and distributional data that are used as input for a biogeographical analysis. Following this principle, Component Compatibility Analysis (CCA) has been developed by dr. Zandee and Brooks Parsimony Analysis (primary BPA, secondary BPA) has been developed by prof. dr. Brooks.
By these methods, widespread and sympatric species are dealt with in a parsimony analysis of the unmodified cladograms. The most parsimonious depiction of all the data is selected as the general area cladogram and the species whose distributions conflict with that pattern are explained a posteriori as extinction or dispersal. The explanations of incongruent distributions of species are obtained by optimizing the data of each monophyletic group on the general area cladogram (CCA and primary BPA) or by duplicating the areas in which the incongruent distributions occur (secondary BPA).
The aim of this project is to assess the properties of a posteriori methods for phylogenetic biogeographical analyses. In close collaboration with prof. dr. Brooks (Zoological Department, University of Toronto) and dr. Zandee (Institute of Biology, Leiden University) the results of this project may be used for further development and implementation of a posteriori methods.
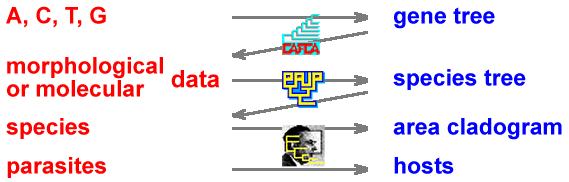
In all these studies, phylogenetic relationships between entities (genes, species, areas or hosts) are obtained from molecular, morphological, biogeographical or parasitological data. These phylogenetic relationships are represented in cladograms. By assuming that the obtained cladograms are hypotheses of gene isolation or speciation events, evolutionary processes are inferred after optimizing molecular, morphological, biogeographical or parasitological data on the cladograms.
The aim of this project is to specify the analogies upon which the application of phylogenetic methodology for comparative studies in (molecular) systematics, historical biogeography and studies of host-parasite associated systems is justified. It is examined whether these analogies in all these different fields hold and if additional steps to methods for obtaining cladograms (by parsimony analysis) are necessary.
This project is performed in close collaboration with prof. dr. D.R. Brooks (Zoological Department, University of Toronto).